

Exercise 1: Search with a sequence to identify the pathogen
Task: Identify the patient's pathogen based on a nucleotide sequence
A common exercise is to identify a pathogen sample based on several methods including culturing and then examination of microscopic features, histopathology, serology, immunoassays, and for bacteria - growth media requirements, and biochemical analysis. More recently DNA sequencing has dramatically sped up and increased the sensitivity of the identification of an organism, as seen with the new COVID-19 PCR tests.
You will use NCBI's BLAST Service to identify the likely organism causing your patient's infection.
Background
A common practice to identify bacterial and fungal pathogens is based on "targeted loci" sequencing & comparison.NCBI's Targeted Loci Project (https://www.ncbi.nlm.nih.gov/refseq/targetedloci/)
This project creates curated BLAST databases which include selected RefSeq records and validated GenBank sequences. Amplification of specific regions with universal primers in bacterial isolates or fungal isolates generate sequences that can be used for comparison with known reference sequences.
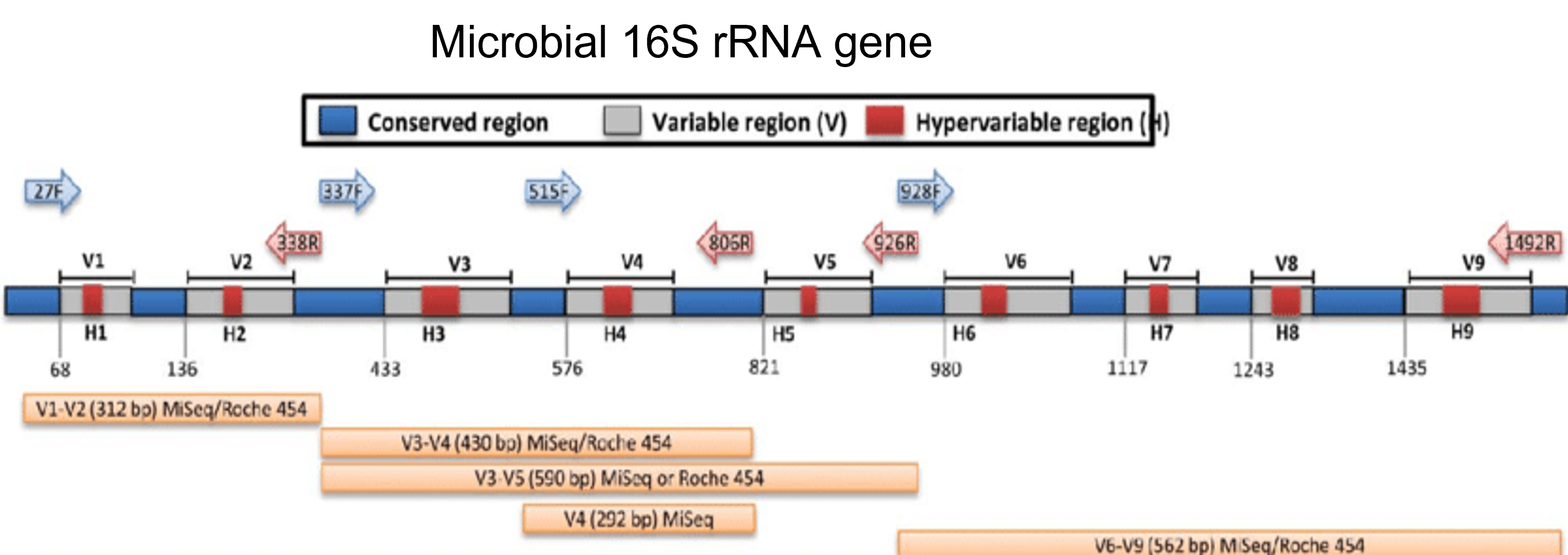
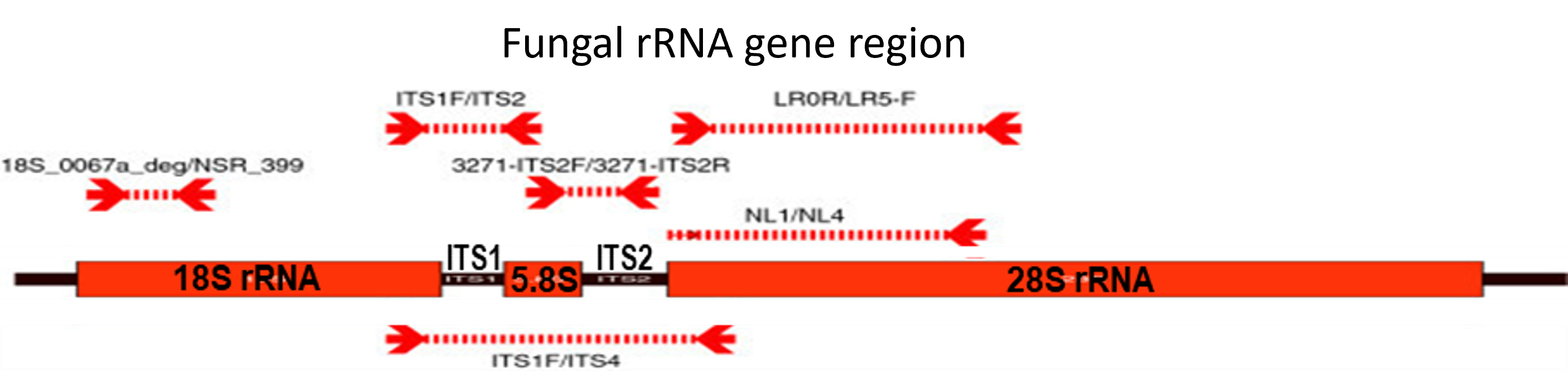
This project creates curated BLAST databases which include selected RefSeq records and validated GenBank sequences. Amplification of specific regions with universal primers in bacterial isolates or fungal isolates generate sequences that can be used for comparison with known reference sequences.
- Bacteria and Archaea: 16S rRNA gene - full length 16S ribosomal RNA sequences that correspond to bacteria and archaea type materials.
- Bacteria and Archaea: 23S rRNA gene - selected complete and near full length sequences
- Fungal genome region:
- 18S (SSU) rRNA gene - regions containing most of the variable V4 region and part of the V5 region
- Internal transcribed spacer (ITS) regions - near full length to complete ITS1, 5.8S gene and ITS2 sequences
- 28S (LSU) rRNA gene - regions containing the hypervariable D1/D2 region
A common practice to identify viral pathogens is by amplification & sequencing of key genomic regions.
NCBI RefSeq Viral Genomes (https://www.ncbi.nlm.nih.gov/genome/viruses/)
The team helps to curate a BLAST database of viral RefSeq genome sequences.
The team helps to curate a BLAST database of viral RefSeq genome sequences.
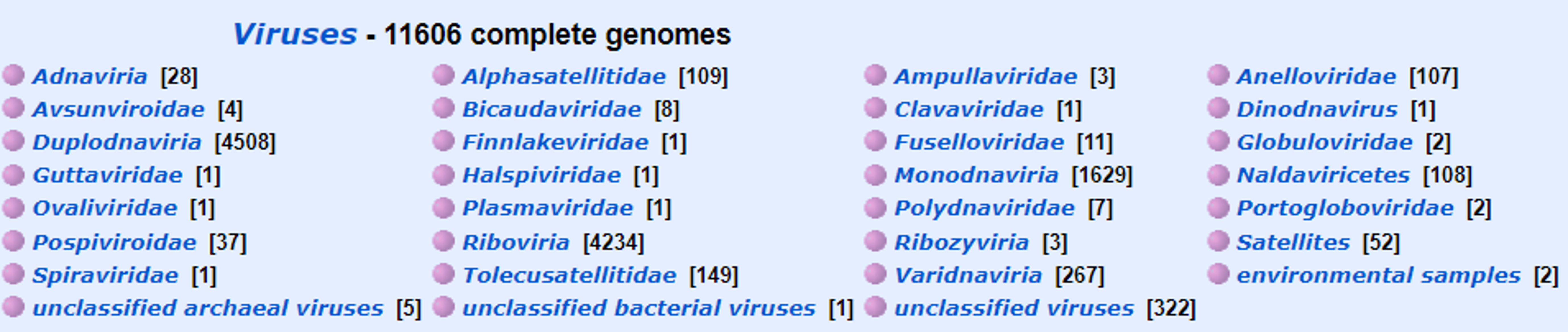
This database will be basis for identifying the source of portions of genomes that are amplified and sequenced.
The specific region or genes that are targeted for sequencing are specific for each viral family, for example:
The specific region or genes that are targeted for sequencing are specific for each viral family, for example:
- Influenza A
- Hemagglutinin (18 subtypes): surface glycoprotein responsible for docking and membrane fusion for entry into host cells
- Neuraminidase (11 subtypes): surface protein promotes release of the virus from the host cell
- HIV
- Integrase portion of the polymerase gene
- Dengue
- 4 serotypes – Non-structural peptide 5 (NS5)
- SARS-CoV-2
- CDC: Nucleocapsid (N) gene
- ORF1ab
Key NCBI Resources for this Exercise
NCBI BLAST - The Basic Local Alignment Search Tool (BLAST) finds regions of local similarity between sequences. The program compares nucleotide or protein sequences to sequence databases and calculates the statistical significance of matches. BLAST can be used to retrieve similar sequences with informative metadata to infer the source organism for the isolate, identify potentially related members of gene families, as well as explore evolutionary or functional relationships between sequences.
Specific BLAST databases for this exercise:
Specific BLAST databases for this exercise:
Your turn: Identify your patient's pathogen!
Pick your case study, click the link to go to that section to start!
Suspected viral infection
6-year-old male with mild non-productive cough, runny nose, sore throat, and headache for 1 week, suddenly developed a fever and rash on his face and trunk. Recent international travel visiting with family in Nigeria. A nasopharyngeal swab sample was obtained and sent out for RT-PCR Viral Nucleoprotein sequencing.
The results come back:
| >Suspected Viral Infection TGGCATCCGAACTCGGTATCACTGCCGAGGATGCAAGGCTTGTTTCAGAGATTGCAATGCATACTACTGAGGACA |
Here's the steps you need to do:
- Go to the BLAST home page (https://blast.ncbi.nlm.nih.gov), then click Nucleotide BLAST.
- Copy/paste the results into the “Enter Query Sequence” box.
- Next to “Database”, click the pull-down menu, select “RefSeq Genome database”.
- In the “Organism” box type “Viruses” and click on the offered “Viruses (taxid:10239)”
- Scroll down and click the BLAST button.
- Let the blast search run and shortly the page will load with the results.
If you need it, you can click here to get to a link for BLAST result page.
- Scroll down and look at the table to see what the highest percentage match is. You can also look at the exact alignments to see the matches by clicking on an organism’s name.
Things to consider
-
- The Descriptions tab of the BLAST report provides a quick view of the results. If a list of close results is returned (which is not uncommon for the 16S rRNA database searchers), Percent Identity is often and important statistic (since the e-value is impacted by match length).
- Confirm identification by looking at selected sequence Alignments.
- You can also select to view all the hits with the MSA viewer (multiple sequence alignment) which will enable more display options to assist sequence comparison.
- Viewing a Distance tree of results provides a quick, BLAST-based phylogenetic tree of the alignments. This is another way to find other sequences that are most similar to your sample.
What do you think your patient has?
Click here to see the answer!

Go to the Take-away Message!
Suspected bacterial infection
19-year-old female, admitted to the hospital from the ER after 2 days of persistent diarrhea and fever. Had recently been to a local restaurant with her biochemistry lab group and ate a whole plate of the daily special - sauteed pea shoots. A poo swab sample was obtained and sent out for RT-PCR microbial 16S rRNA sequencing.
The results come back:
| >Suspected Bacterial Infection CTGATGGAGGGGGATAACTACTGGAAACGGTGGCTAATACCGCATAACGTCGCAAGACCAAAGAGGGGGACCTTCGGGCCTCTTGCCATCAGATGTGCCCAGATGGGATTAGCTAGTTGGTGAGGT |
Here's the steps you need to do:
- Go to the BLAST home page (https://blast.ncbi.nlm.nih.gov), then click Nucleotide BLAST.
- Copy/paste the results into the “Enter Query Sequence” box.
- Next to “Database”, click “rRNA/ITS databases” and then click the pull-down menu to select “16S ribosomal RNA sequences”.
- Scroll down and click the BLAST button.
- Let the blast search run and shortly the page will load with the results.
If you need it, you can click here to get to a link for BLAST result page.
- Scroll down and look at the table to see what the highest percentage match is. You can also look at the exact alignments to see the matches by clicking on an organism’s name.
Things to consider
-
- The Descriptions tab of the BLAST report provides a quick view of the results. If a list of close results is returned (which is not uncommon for the 16S rRNA database searchers), Percent Identity is often and important statistic (since the e-value is impacted by match length).
- Confirm identification by looking at selected sequence Alignments.
- You can also select to view all the hits with the MSA viewer (multiple sequence alignment) which will enable more display options to assist sequence comparison.
- Viewing a Distance tree of results provides a quick, BLAST-based phylogenetic tree of the alignments. This is another way to find other sequences that are most similar to your sample.
What do you think your patient has?
Click here to see the answer!

Go to the Take-away Message!
Suspected fungal infection
50-year-old male, presenting with shortness of breath, low-grade fever, and an inability to stand without assistance - history of diabetes mellitus and renal insufficiency with recent hemodialysis treatment in a healthcare facility in India (visiting family). An arteriovenous treatment site appeared infected. A blood sample was taken and sent to the lab for a fungal Rapid PCR diagnostic test.
The results come back:
| >Suspected Fungal Infection CAGCGAAATGCGATACGTAGTATGACTTGCAGACGTGAATCATCGAATCTTTGAACGCACATTGCGCCTTGGGGTATTCCCCAAGGCATGCCTGTT |
Here's the steps you need to do:
- Go to the BLAST home page (https://blast.ncbi.nlm.nih.gov), then click Nucleotide BLAST.
- Copy/paste the results into the “Enter Query Sequence” box.
- Next to “Database”, click “rRNA/ITS databases” and then click the pull-down menu to select “Internal transcribed spacer region (ITS)”.
- Scroll down and click the BLAST button.
- Let the blast search run and shortly the page will load with the results.
If you need it, you can click here to get to a link for BLAST result page.
- Scroll down and look at the table to see what the highest percentage match is. You can also look at the exact alignments to see the matches by clicking on an organism’s name.
Things to consider
-
- The Descriptions tab of the BLAST report provides a quick view of the results. If a list of close results is returned (which is not uncommon for the 16S rRNA database searchers), Percent Identity is often and important statistic (since the e-value is impacted by match length).
- Confirm identification by looking at selected sequence Alignments.
- You can also select to view all the hits with the MSA viewer (multiple sequence alignment) which will enable more display options to assist sequence comparison.
- Viewing a Distance tree of results provides a quick, BLAST-based phylogenetic tree of the alignments. This is another way to find other sequences that are most similar to your sample.
What do you think your patient has?
Click here to see the answer!
 NOTE: In the BLAST results, it doesn't show Candida auris - but instead lists [Candida] auris. Why is this?
NOTE: In the BLAST results, it doesn't show Candida auris - but instead lists [Candida] auris. Why is this?The square brackets indicate that this name may have been misclassified and is a temporary name and awaiting formal renaming. Here's an explanation of this which actually describes the [Candida] auris situation.
Because all NCBI records for this organism will use the NCBI Taxonomy designation, you should use the square brackets (i.e. [Candida] auris) when doing your searches.
Take-away message!
To compare and identify your viral isolate sequence:
- Use BLAST and select the database "RefSeq Genomes" and filter for Viral sequences.
- Use BLAST and select the "16S ribosomal RNA (rRNA) - Targeted Loci".
- Use BLAST and select the database "Transcribed Spacer (ITS) region - Targeted Loci".
For more advanced work....
Learn more about BLAST
-
- BLAST Help Documents
- NCBI BLAST YouTube Video Playlist - including:
- An NCBI Workshop: "Using Web BLAST Effectively (October 18, 2022)"
- An NCBI Workshop: "An Update on NCBI BLAST and Other Sequence Analysis Tools (January 25, 2022)"
Create your own primers or probes
Last Reviewed: July 27, 2023